Every MI scene graph must have the following nodes that control the manner in which the chemical system data is rendered:
The ChemUI node provides an interface that allows a user to change the values of fields in the ChemDisplayParam node thus changing the display.
This chapter describes how to set the values in all of these nodes in the following sections:
“Controlling the Color of Atoms, Bonds, and Labels” discusses the ChemColor node.
“Controlling the Atom Radii” discusses the ChemRadii node.
“Changing Display Parameters” discusses the ChemDisplayParam node.
“Using ChemUI” discusses how to use a ChemUI node.
The ChemColor node controls the colors of atoms, bonds, and labels. The color values are maintained in four tables within the ChemColor node; one each for atoms, bonds, atom labels, and bond labels. These tables are named:
Atoms, bonds, and labels can all be different colors. In practice, however, it is often useful for labels of atoms to be the same color as their atoms, and labels of bonds to be the same color as their bonds. It is also common to color bonds the same colors as the atoms that are joined by the bonds. The binding fields in ChemColor specify whether all items of a given type are colored the same, individually, or according to an index that references an entry in one of the color tables.
The following figure shows the display of miApp, in which the bonds are the same color as their atoms.
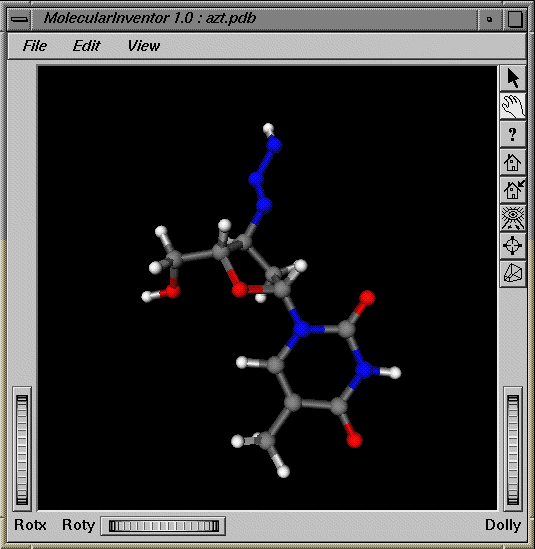
If atom labels are the same color as their atoms, a color table for atom labels is not used; methods requesting the color of atom labels are diverted instead to the atom color table. The same is true for bond labels: there is no need to maintain a bond label color table if the bond labels are the same color as the bonds. Likewise, if the bonds are the same color as the atoms which they join, there is no need to maintain a bond color table.
Each of the color binding fields in ChemColor is of type SoSFEnum, as shown in the header file, ChemColor.h:
SoSFEnum atomColorBinding SoSFEnum bondColorBinding SoSFEnum atomLabelColorBinding SoSFEnum bondLabelColorBinding |
SoSFEnum is a single-value field that contains an enumerated type stored as an integer. The following example shows how to set the various color binding values:
atomColorBinding.setValue(ChemColor::ATOM_PER_ATOM); bondColorBinding.setValue(ChemColor::BOND_PER_ATOM_HALF_BONDED); atomLabelColorBinding.setValue(ChemColor::ATOM_LABEL_PER_ATOM_LABEL_INDEXED); bondLabelColorBinding.setValue(ChemColor::BOND_LABEL_PER_BOND_INDEXED); |
 | Note: You must precede all field values with ChemColor::. |
“Coloring Atoms,” “Coloring Bonds,” “Coloring Atom Labels,” and “Coloring Bond Labels” describe the different color bindings you can specify for atoms, bonds, and labels.
Molecular Inventor provides default atomColor, bondColor, atomLabelColor, and bondLabelColor tables. The table entries are based on the default settings of the color binding fields. The number of entries in a color table depends on the values used for the color bindings.
The atomColor table assigns colors to atoms according to the value of the atomColorBinding field in ChemColor. The following table describes the possible values of atomColorBinding.
Table 4-1. atomColorBinding Values
Value of atomColorBinding | Description |
|---|---|
ATOM_OVERALL | The first entry in the atomColor table is used as the color value for all atoms. |
ATOM_PER_ATOM | Each atom has its own entry in the atomColor table; there must be as many entries in the table as there are atoms. |
ATOM_PER_ATOM_INDEXED | The value returned by the getAtomIndex() method in ChemBaseData is used as an index into the atomColor table. |
ATOM_DEFAULT | The default is ATOM_PER_ATOM_INDEXED. |
The default atomColor table contains 104 entries and assumes that atomic numbers are used to index into the table. The CPK color scheme is used in the table.
The color of bonds is set by the bondColorBinding field. Bonds can all be colored differently, the same, or the same as the atoms joined by the bond. In the last case, the color values in the atomColor table are used for the bonds.
The following table describes the possible values of bondColorBinding.
Table 4-2. bondColorBinding Values
Value of bondColorBinding | Description |
|---|---|
BOND_OVERALL | The first entry in the bondColor table is used as the color value for all bonds. |
BOND_PER_BOND | Each bond has its own entry in the bondColor table; there must be as many entries in the table as there are bonds. |
BOND_PER_BOND_INDEXED | The value returned by the getBondIndex() method in ChemBaseData is used as an index into the bondColor table. |
BOND_PER_ATOM | This value colors the bonds using the colors of the atoms that comprise the bond. The bondColor table is not used. In the case where the bond is comprised of two different types of atoms, the bond is displayed using two different colors. In BOND_PER_ATOM, these colors are blended along the length of the bond. |
BOND_PER_ATOM_HALF_BONDED | This value acts similarly to BOND_PER_ATOM except that the two colors of the bond are not blended; instead, one half of the bond is drawn using the color of one atom and the other half of the bond is drawn using the color of the other atom, as shown in Figure 4-1. |
BOND_DEFAULT | The default is BOND_PER_ATOM_HALF_BONDED. |
Since the default value, BOND_DEFAULT, colors bonds according to the atomColor table, the default bondColor table contains only one entry as a place holder.
The atomLabelBinding field specifies the method used to color atom labels. Atom labels can be colored according to their own color table, atomLabelColor, or according to the color of the atom the label is associated with. The following table describes the possible values of atomLabelBinding.
Table 4-3. atomLabelBinding Values
Value of atomLabelBinding | Description |
|---|---|
ATOM_LABEL_OVERALL | The first entry in the atomLabelColor table is used as the color value for all atom labels. |
ATOM_LABEL_PER_ATOM | The atom labels are colored the same as the atoms they describe. The atomColor table and atomColorBinding are used to determine the color of the atom label. |
ATOM_LABEL_PER_ATOM_LABEL | Each atom label has its own entry in the atomLabelColor table; there must be as many entries in the table as there are atoms. |
ATOM_LABEL_PER_ATOM_LABEL_INDEXED | The value returned from the getAtomIndex() method in ChemBaseData is used as an index into the atomLabelColor table. |
ATOM_LABEL_DEFAULT | The default is ATOM_LABEL_OVERALL. |
The default atomLabelColor table contains one entry: the color white, (1, 1, 1).
The bondLabelBinding field specifies the method used to color bond labels. Bond labels can be colored according to their own color table, bondLabelColor, or according to the bond the label is associated with. The following table describes the possible values of bondLabelBinding.
Table 4-4. bondLabelBinding Values
Value of bondLabelBinding | Description |
|---|---|
BOND_LABEL_OVERALL | The first entry in the bondLabelColor table is used as the color value for all bond labels. |
BOND_LABEL_PER_BOND | The bond labels are colored the same as the bonds they describe. The bondColor table is used and there must be as many entries in the bondColor table as there are bonds. |
BOND_LABEL_PER_BOND_INDEXED | The value returned by the getBondIndex() method in ChemBaseData is used as an index into the bondLabelColor table. |
BOND_LABEL_PER_BOND_LABEL | The bond labels are colored according to the entries in the bondLabelColor table. There must be as many entries in the bondLabelColor table as there are bonds. |
BOND_LABEL_PER_BOND_LABEL_INDEXED | The value returned from the getBondIndex() method in ChemBaseData is used as an index into the bondLabelColor table. |
BOND_LABEL_DEFAULT | The default is BOND_LABEL_OVERALL. |
The default bondLabelColor table contains one entry: the color white, (1, 1, 1).
When atoms are displayed as spheres, the radii of the spheres are obtained from the atomRadii table in the ChemRadii node. Atom radii can all be the same size, can be set to different sizes individually, or set according to the atom's index number, as returned by getAtomIndex().
The atomRadii table assigns radii to atoms according to the value of the atomRadiiBinding field in the ChemRadii node. This field is of type SoSFEnum, which is a single-value field that contains an enumerated type stored as an integer. The following example shows how to set the value of the atomRadiiBinding field:
atomRadiiBinding.setValue(ChemRadii::RADII_PER_ATOM_INDEXED); |
| Note: You must precede the radii values with ChemRadii::. |
The following table describes the possible values of atomRadiiBinding.
Table 4-5. atomRadiiBinding Values
Value of atomColorBinding | Description |
|---|---|
RADII_OVERALL | The first entry in the atomRadii table is used as the radius for all atoms. |
RADII_PER_ATOM | Each atom has its own entry in the atomRadii table; there must be as many entries in the table as there are atoms. |
RADII_PER_ATOM_INDEXED | The value returned by the getAtomIndex() method in ChemBaseData is used as an index into the atomRadii table. |
RADII_DEFAULT | The default is RADII_PER_ATOM_INDEXED. |
The default atomRadii table contains 104 entries and assumes that the numbers used to index into the table correspond to atomic numbers.
 | Tip: All of the entries of the atomRadii table can easily be scaled by setting the atomRadiiScaleFactor field in the ChemDisplayParam node to a value other than 1.0. |
The ChemDisplayParam node contains many fields, each of which affects the rendering of the chemical system. Some of the parameters include, for example, whether the system is rendered as a wireframe or as spheres and cylinders, or whether or not fog is used to enhance the sense of depth.
The fields in the ChemDisplayParam node are of various types, each of which is a class. All of the classes contain the setValue() method, which you use to set display parameter values. The only fields that you have to set are those you want to change from the default value. The following sections explain each of these fields and their possible values.
The displayStyle field sets the rendering style of the atoms and bonds, for example, the atoms and bonds can appear as lines, or the atoms can appear as spheres and the bonds as cylinders.
The displayStyle field is equal to one of the values in the DisplayBinding enum shown in the following table.
Table 4-6. DisplayBinding Values
DisplayBinding Values | Description |
|---|---|
DISPLAY_NONE | Nothing is rendered. |
Draws atoms as spheres. | |
Draws bonds as cylinders. Any non-bonded atoms are drawn according to the value of the nonBondedAtomStyle field. | |
Draws atoms as scaled spheres and bonds as cylinders. | |
Draws bonds as lines. Any non-bonded atoms are drawn according to the value of the nonBondedAtomStyle field. | |
Draws atoms as scaled spheres and bonds as lines. | |
DISPLAY_DEFAULT | Default is DISPLAY_WIREFRAME. |
The showHydrogens field is a Boolean value. Setting the field to FALSE turns off the display of hydrogen atoms; setting the field to TRUE, the default, turns on the display of hydrogen atoms.
| Note: showHydrogens toggles the display of atoms whose atomic number is 1. If you set the atomic number of another atom to 1, showHydrogens toggles the display of this atom. |
When atoms are rendered as spheres, the following fields control aspects of the spheres.
The atomRadiiScaleFactor field value scales the size of the atomic radii set by the ChemRadii node. Entries in the atomRadii table are multiplied by this value when the displayStyle is ChemDisplayParam::DISPLAY_CPK. The default is 1.0, no scaling.
The entries in the atomRadii table in the ChemRadii node are multiplied by the ballStickSphereScaleFactor field value when the display style is ChemDisplayParam::DISPLAY_BALLSTICK or ChemDisplayParam::DISPLAY_BALLWIRE. The default scale factor is 0.2.
The atomSphereComplexity field value, which lies between 0.0 and 1.0, determines the number of triangles used when rendering a sphere that represents an atom. This value has the same effect as the value used in a SoComplexity node.
The atomSphereDisplayStyle field determines whether atoms are displayed as hemispheres that always face the camera or as full spheres. The default is hemispheres since they often render faster. The following table shows the valid values for the atomSphereDisplayStyle field.
Table 4-7. atomSphereDisplayStyle Values
atomSphereDisplayStyle Values | Description |
|---|---|
ATOMSPHERE_HEMISPHERES | Renders atoms using hemispheres. |
ATOMSPHERE_FULLSPHERES | Renders atoms using full spheres. |
ATOMSPHERE_DEFAULT | Default is ATOMSPHERE_HEMISPHERES. |
When bonds are displayed as lines, the following fields control various aspects of how the lines are rendered.
The showMultipleBonds field is a Boolean value. Setting the value to FALSE, causes all bonds to be rendered as single lines; setting the value to TRUE, the default, causes multiple lines to be rendered when necessary, as in the case of a double bond. This field is checked only when the displayStyle is ChemDisplayParam::DISPLAY_WIREFRAME or ChemDisplayParam::DISPLAY_BALLWIRE.
The multipleBondSeparation field sets the spacing between the multiple lines rendered when showMultipleBonds is set to TRUE. The default is 0.15 Angstroms.
The bondWireframeAntiAlias field is a Boolean value. Setting the field to FALSE, the default, turns off antialiasing of the lines; setting the field to TRUE turns on antialiasing.
The bondWireframeLineWidth field value, which must be greater than 0.0, sets the width of the lines used. The default is 1.0.
When bonds are displayed as cylinders, the following fields control various aspects of the cylinders.
The bondCylinderRadius field sets the radius of the cylinder that represents the bonds. The default is 0.15 Angstroms.
The bondCylinderComplexity field value, which lies between 0.0 and 1.0, determines the number of triangles used when rendering the cylinder. This value has the same effect as the value used in a SoComplexity node.
The bondCylinderDisplayStyle field determines whether or not caps are rendered on the ends of cylinders and whether that cap is flat or rounded. The bondCylinderDisplayStyle field is equal to one of the values in the BondCylinderBinding enum shown in the following table.
Table 4-8. bondCylinderDisplayStyle Values
bondCylinderDisplayStyle Values | Description |
|---|---|
BONDCYLINDER_NOCAP | Does not cap the cylinders. |
BONDCYLINDER_FLATCAP | Puts flat caps on the cylinders. |
BONDCYLINDER_ROUNDCAP | Puts rounded caps on the cylinders. |
BONDCYLINDER_DEFAULT | Default is BONDCYLINDER_NOCAP. |
The following fields are used when the displayStyle field is set to either ChemDisplayParam::DISPLAY_STICK or ChemDisplayParam::DISPLAY_WIREFRAME. In theses cases, atoms are not displayed. The following fields make it possible, however, to select atoms and to display atoms not contained in bonds.
The atomPickRadius field defines the radius of a sphere around the ends of a bond within which a user can click to select an atom. If the value of atomPickRadius is 0.20, for example, an atom is picked if the user clicks within 0.2.0 Angstroms of the end of the bond. The default value is 0.20.
The nonBondedAtomStyle field value determines how atoms not contained in bonds are displayed when displayStyle is either ChemDisplayParam::DISPLAY_WIREFRAME or ChemDisplayParam::DISPLAY_STICK. The nonBondedAtomStyle field is equal to one of the values in the NonBondedAtomBinding enum shown in the following table.
Table 4-9. nonBondedAtomStyle Values
nonBondedAtomStyle Values | Description |
|---|---|
NONBONDED_ATOM_NONE | Does not display non-bonded atoms. |
NONBONDED_ATOM_BITMAP | Displays atoms using a bitmap. |
NONBONDED_ATOM_DEFAULT | Default is NONBONDED_ATOM_BITMAP. |
If the NONBONDED_ATOM_BITMAP value is chosen, the bitmap is set by the nonBondedAtomBitmap field and is rendered at the location of every atom not contained in a bond. The bitmap is stored as an array of unsigned bytes in the nonBondedAtomBitmap field. The default bitmap is an “X”.
The nonBondedAtomBitmapHeight and nonBondedtAtomBitmapWidth fields specify the height and width of the bitmap. For further information about using bitmaps, see the discussion on “Highlighting Selected Items.”
The following fields set whether atom labels are displayed, and, if so, the justification of the label with respect to the coordinates of an atom. The atomLabelIndex field in ChemDisplay controls which atom labels are actually displayed.
The showAtomLabels field is a Boolean value. Setting the field to FALSE, the default, turns off the display of atom labels; setting the field to TRUE turns on the display of atom labels.
The atomLabelLeftRightJustification field determines where an atom's label is rendered horizontally relative to the atom's coordinates. For example, ChemDisplayParam::LABEL_LR_LEFT positions the label so that its left edge is at the atom's coordinates.
The atomLabelLeftRightJustification field is equal to one of the values in the LabelLeftRightJustificationBinding enum shown in the following table.
Table 4-10. atomLabelLeftRightJustification Values
atomLabelLeftRightJustification Values | Description |
|---|---|
LABEL_LR_LEFT | The label is left-justified with respect to the coordinates. |
LABEL_LR_RIGHT | The label is right-justified with respect to the coordinates. |
LABEL_LR_CENTER | The label is centered with respect to the coordinates. |
LABEL_LR_DEFAULT | The default is LABEL_LR_LEFT. |
The atomLabelTopBottomJustification field determines where an atom's label is rendered vertically relative to the atom's coordinates. For example, ChemDisplayParam::LABEL_LR_TOP positions the label so that its top edge is at the atom's coordinates.
The atomLabelTopBottomJustification field is equal to one of the values in the LabelTopBottomJustificationBinding enum shown in the following table.
Table 4-11. atomLabelTopBottomJustification Values
atomLabelTopBottomJustification Values | Description |
|---|---|
LABEL_TB_TOP | The label is top-justified with respect to the coordinates. |
LABEL_TB_BOTTOM | The label is bottom-justified with respect to the coordinates. |
LABEL_TB_MIDDLE | The label is top-bottom centered with respect to the coordinates. |
LABEL_TB_DEFAULT | The default is LABEL_TB_BOTTOM. |
The atomLabelString field sets the string used for the atom's label. In addition to supplying your own text, you can use any combination of the variables in the following table to specify the string.
Table 4-12. String Variables for Atoms
Variable | Description |
|---|---|
%a | The atom's symbol. |
%i | The atom's ID. |
%e | The atom's atomic number. |
For example, setting the label to
atomLabelString.setValue(“Moll_%a_%i”); |
creates a label similar to Moll_C_31. The default is %a.
The following fields set whether bond labels should be displayed, and if so, the justification of the label with respect to the midpoint of the bond. The bondLabelIndex field in ChemDisplay controls which bond labels are actually displayed.
The showBondLabels field is a Boolean value. Setting the field to FALSE, the default, turns off the display of bond labels; setting the field to TRUE turns on the display of bond labels.
The bondLabelLeftRightJustification field determines where a bond's label is rendered horizontally relative to the midpoint of the bond, for example, ChemDisplayParam::LABEL_LR_LEFT positions the label so that its left edge is at the midpoint of the bond. The bondLabelLeftRightJustification field is equal to one of the values in the LabelLeftRightJustificationBinding enum shown in the following table.
Table 4-13. bondLabelLeftRightJustification Values
bondLabelLeftRightJustification Values | Description |
|---|---|
LABEL_LR_LEFT | Label is left-justified with respect to the midpoint. |
LABEL_LR_RIGHT | Label is right-justified with respect to the midpoint. |
LABEL_LR_CENTER | Label is left-right centered with respect to the midpoint. |
LABEL_LR_DEFAULT | Default is LABEL_LR_LEFT. |
The bondLabelTopBottomJustification field determines where a bond's label is rendered vertically relative to the midpoint of the bond, for example, ChemDisplayParam::LABEL_TB_TOP positions the label so that its top edge is at the midpoint of the bond. The bondLabelTopBottomJustification field is equal to one of the values in the LabelTopBottomJustificationBinding enum shown in the following table.
Table 4-14. bondLabelTopBottomJustification Values
bondLabelTopBottomJustification Values | Description |
|---|---|
LABEL_TB_TOP | The label is top-justified with respect to the midpoint. |
LABEL_TB_BOTTOM | The label is bottom-justified with respect to the midpoint. |
LABEL_TB_MIDDLE | The label is top-bottom centered with respect to the midpoint. |
LABEL_TB_DEFAULT | Default is LABEL_TB_BOTTOM. |
The bondLabelString field sets the string used for the bond's label. In addition to supplying your own text, you can use any combination of the variables in the following table to specify the string.
Table 4-15. String Variables for Bonds
Variable | Description |
|---|---|
%d | The bond's length. |
%fx | The bond's “from” atom. |
%tx | The bond's “to” atom. |
In this table, x is one of the values of atomLabelString, shown in Table 4-12. Every bond has a “from” and “to” atom which specifies the atoms which the bond connects. The x is the means of identifying these atoms. For example, if you set the label to
bondLabelString.setValue(“%fi-%ti:%d”); |
the i stands for, according to Table 4-12, the atom's ID. So, a bond label 5-13:1.5400, is read as follows: 5 is the atom ID of the “from” atom, 13 is the atom ID of the “to” atom, and 1.5400 is the length of the bond.
The fontName field sets the font used when rendering atom and bond labels. The available fonts are listed in /usr/lib/DPS/DPSFonts.upr.
The fontSize field sets the font size used when rendering atom and bond labels.
There are various ways in which an application can implement the selection of atoms, bonds, and labels. The following fields specify how selected items are highlighted.
The highlightStyle field determines how selected atoms, bonds, and labels are rendered. The highlightStyle field is equal to one of the values in the HighlightBinding enum shown in the following table.
Table 4-16. highlightStyle Values
highlightStyle Values | Description |
|---|---|
HIGHLIGHT_NONE | Does not highlight selected items. |
HIGHLIGHT_EMISSIVE | Highlights by setting the emissive color. |
HIGHLIGHT_DIFFUSE | Highlights by setting the diffuse color. |
HIGHLIGHT_DRAWSTYLE | Highlights by changing the draw style. |
HIGHLIGHT_DEFAULT | Default is HIGHLIGHT_EMISSIVE. |
When the highlightStyle field is set to ChemDisplayParam::HIGHLIGHT_EMISSIVE or ChemDisplayParam::HIGHLIGHT_DIFFUSE, the highlightColor field controls which color to use for the diffuse color or emissive color of the atom, bond, or label. The color specification is in the RGB format, for example, 0 1 0 sets the color to pure green.
A diffuse color is an object's base color. An emissive color is the color of the light produced by an object. For more information about these colors, see the section on material nodes in The Inventor Mentor.
If the highlightStyle field is set to ChemDisplayParam::HIGHLIGHT_DRAWSTYLE,
all selected atoms and bonds displayed as spheres and cylinders are rendered using unfilled triangles.
the highlightFontName field sets the font used when rendering atom and bond labels that have been selected.
The available fonts are listed in /usr/lib/DPS/DPSFonts.upr. The highlightFontSize field sets the font size for the selected labels when the highlightStyle field is set to ChemDisplayParam::HIGHLIGHT_DRAWSTYLE.
if bonds are displayed as lines, selected bonds are displayed using a stipple pattern. highlightStippleFactor and highlightStipplePattern control the manner in which the stippling is performed.
The highlightStippleFactor field value specifies a multiplier for each bit in the line stipple pattern. If the factor is 3, for example, each bit in the pattern is used three times before the next bit in the pattern is used. The range of highlightStippleFactor field values is between 1.0 and 256.0; the default is 1.0.
The highlightStipplePattern field value specifies a 16-bit integer whose bit pattern determines which fragments of a line are drawn when the line is rasterized. Bit zero is used first. The default pattern is 0x0F0F.
The highlightStippleFactor and highlightStipplePattern field values are used as arguments to the OpenGLR function glLineStipple().
The highlightAtomStyle field determines how selected atoms are rendered when displayStyle is either ChemDisplayParam::DISPLAY_WIREFRAME or ChemDisplayParam::DISPLAY_STICK. The highlightAtomStyle field is equal to one of the values in the HighlightAtomBinding enum shown in the following table.
Table 4-17. highlightAtomStyle Values
highlightAtomStyle Values | Description |
|---|---|
HIGHLIGHT_ATOM_NONE | Does not highlight selected atoms. |
HIGHLIGHT_ATOM_BITMAP | Highlights atoms using a bitmap. |
HIGHLIGHT_ATOM_BBOX | Highlights atoms using a bounding box. |
HIGHLIGHT_ATOM_DEFAULT | The default is HIGHLIGHT_ATOM_BITMAP. |
If ChemDisplayParam::HIGHLIGHT_ATOM_BITMAP is chosen, the bitmap is set by the highlightAtomBitmap field and is rendered at the location of every selected atom. The bitmap is stored as an array of unsigned bytes in the highlightAtomBitmap field. The default bitmap is an X. The highlightAtomBitmapHeight and highlightAtomBitmapWidth fields specify the height and width of the bitmap.
The following code sample shows how these three fields are used by the ChemDisplay node.
xbmap = cdp->highlightAtomBitmap.getValues(0); xbmapWidth = cdp->highlightAtomBitmapWidth.getValue(); xbmapHeight = cdp->highlightAtomBitmapHeight.getValue(); xbmapWidth2 = xbmapWidth * 0.5; xbmapHeight2 = xbmapHeight * 0.5; glPixelStorei(GL_UNPACK_ALIGNMENT, 1); glBitmap(xbmapWidth, xbmapHeight, xbmapWidth2, xbmapHeight2, (GLfloat)xbmapWidth, 0.0, xbmap); |
cdp is a pointer to a ChemDisplayParam node. You should not include this code in your application. This example is presented only to show how the information in the highlightAtomBitmap fields is used so that you can more easily construct an appropriate bitmap.
Fog is the name used in OpenGL and Open Inventor programming to describe various “atmospheric effects.” The most often-used effect in molecular graphics is that of depth-cueing which makes objects fade into the distance. This type of effect is achieved with ChemDisplayParam::FOG_HAZE.
For more information, see the section on “Blending, Anti-aliasing, and Fog” in the OpenGL Programming Guide.
You can also use an Open Inventor SoEnvironment node to control atmospheric effects. The fields in ChemDisplayParam that control atmospheric effects are provided as a convenience so that all rendering settings can be maintained by the ChemDisplayParam node.
The fogType field is equal to one of the values in the FogBinding enum, shown in the following table.
fogType Values | Description |
|---|---|
FOG_NONE | No fog. |
A linear increase in opacity with distance. | |
An exponential increase in opacity with distance. | |
An exponential squared increase in opacity with distance. | |
FOG_DEFAULT | The default is FOG_NONE. |
When a the fogType field is other than ChemDisplayParam::FOG_NONE, the fogColor field sets the color which is blended with each point in an object to create the atmospheric effect. The color specification is in the RGB format, for example, 0 1 0 sets the color to pure green. The default is black, 0 0 0.
When the fogType field is ChemDisplayParam::FOG_FOG or ChemDisplayParam::FOG_SMOKE, the fogDensity field sets the density of the fog, which is the blending factor of objects fading into the fog. The default is 1.0, where 0.0 is zero density.
When the fogType field is ChemDisplayParam::FOG_HAZE, the fogStart field sets the distance from the viewer's point of view where the fog should start taking effect. The default value is 0.0.
When the fogType field is other than ChemDisplayParam::FOG_NONE, the fogVisibility field sets the distance from the viewer's point of view where the fog should stop taking effect. The default value is 0.0, which specifies that the far clipping plane should be used.
The clipPlane field holds multiple instances of the Open Inventor SoClipPlane class thereby allowing multiple clipping planes to be maintained in the ChemDisplayParam node. It is not required that instances of SoClipPlane be stored in the clipPlane field; the instances of SoClipPlane can instead appear as nodes in the scene graph. The clipPlane field is provided as a convenience so that all rendering settings can be maintained by the ChemDisplayParam node.
For more information about clipping planes, see the reference page for SoClipPlane.
A ChemUI node provides a simple user interface that enables a user to change the display of a chemical system. Since this interface is part of a scene graph, not the viewer, it can appear in any Inventor-based application, even those that have no knowledge of chemistry data.
The following figure shows the ChemUI interface.
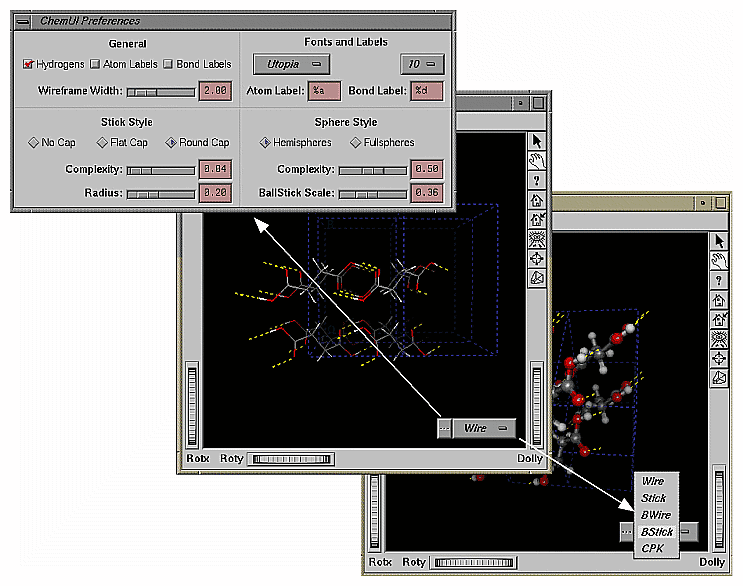
The widget for the ChemUI interface is a two-part button. The left side of the button, labeled with an ellipsis (...), displays the ChemUI Preferences window. In it, you can control, among other things, the width of the line used in wireframe renderings, the style of cylinders used to represent a bond, the font and point size of atom and bond labels, and the number of triangles used to render the spheres and cylinders representing atoms and bonds.
The right side of the ChemUI button displays a pulldown menu which gives the user an option of molecular display styles, including wireframe and BStick (ball and sticks). The current display style appears on the right side of the ChemUI button.